本文章为原创,版权归作者刘锦程所有,文章转载请先取得作者的同意,非常欢迎转发文章链接!严禁以任何方式挪用本文内容,用于以盈利为目的各种活动。
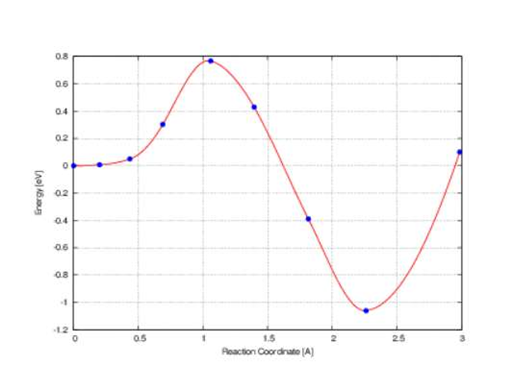
问题一:CI-NEB计算得到的中间有点的能量比初态和末态都要低。
解:这时候有两种可能,
一是在初态和末态之间还存在至少一个极小点;
二是,初态和末态本就不是极小点,需要用更严格的精度重新继续计算初态和末态。
问题二:CI-NEB计算开始出现极大的原子受力,比如10 eV/Angstrom 以上
解:这个原因肯定是差点的初始结构不合理,办法用是VTST作者自己搞的idpp方法,许楠博士提供了idpp.py脚本使用pymatgen实现了这个功能,需要调用pymatgen和pymatgen_diffusion
1 | from pymatgen.core import Structure |
用法:
1 | python3 ./idpp.py ini/POSCAR fin/POSCAR 4 |
或者把不合理的POSCAR下载下来,人工调整这些不合理结构。
问题三:DIMER计算一直不收敛,过程中NEWMODECAR的振动模已经完全偏离了我们预想的方式。
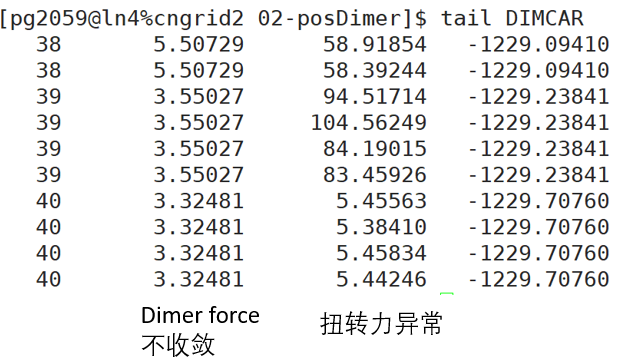
解:是否出现这种问题可以运行用dimmode.pl脚本,把生成的dimmode.xyz放到Jmol里看轨迹,振动方向是不是已经完全偏离我们设想的过渡态。
这时候有可能是力的计算精度不好和初始结构不可理造成的,解决方法有几种,比如可以先固定住其他和反应不相干的原子找过渡态,然后在此基础上再正常固定原子算。
问题四:已经接近收敛,但是很久受力不能达到收敛标准EDIFFG。
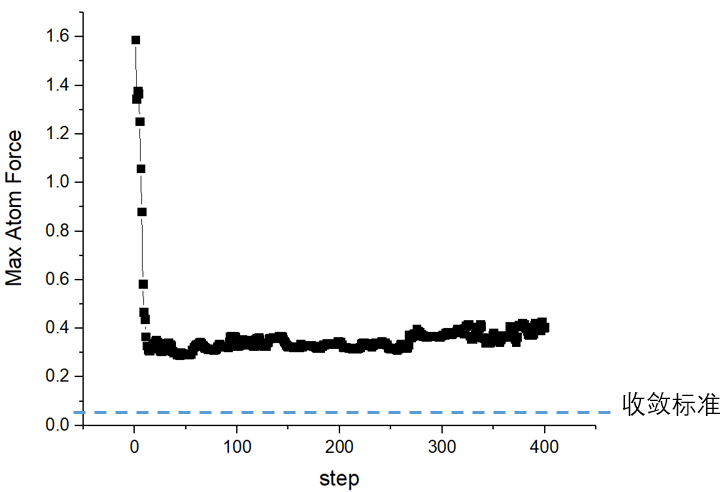
解:
一可能是力的精度不够,用PREC=accurate,EDIFF=1E-7,
二可能是过渡态的优化算法不合适,尝试IOPT=1或2或7,或者IOPT=0,用vasp自带的DIIS优化。
三可能是力的收敛标准过于严格,EDIFFG = 0.02或0.03对于过渡态来说就可以了。
最有效的解决方案是使用我在 解决VASP计算SCF不收敛的方法总结博文中提供的粗收敛+精收敛方案。
问题五:没有虚频或者有两个以上的虚频。
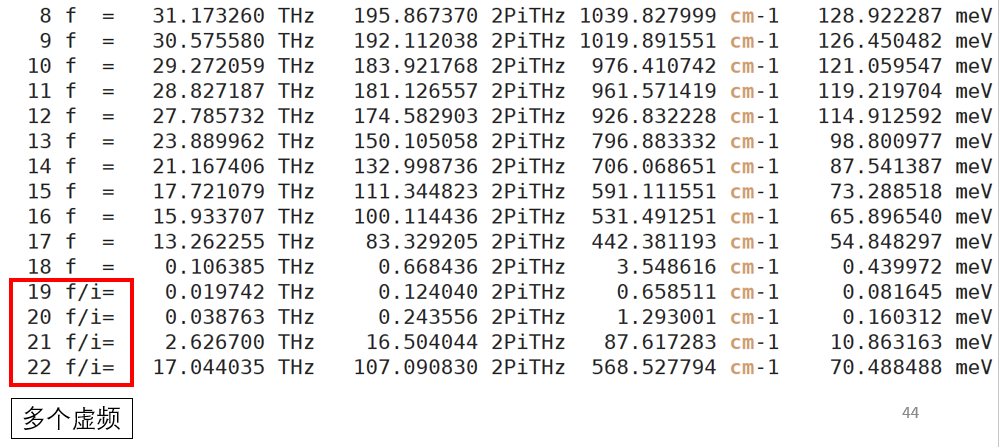
解:
首先我们要确保频率计算的精度够高,至少要和过渡态的EDIFF要求一样。
如果没有虚频,那计算肯定是错的,最可能的还是力的精度不够。算法决定了一般用CI-NEB和Dimer正常结束不会碰到没有虚频的情况。
如果有多个虚频,这个是常见问题,也可能是力的精度不够,这时候可以把结构向着较小的哪个虚频的方向做微小的位移重新作为初始结构计算,俗称消虚频。
问题六:插点结构非常混乱
要检查初态和末态的原子顺序是不是一一对应的!这个是新手的常见错误,所以在计算初态和末态的时候就要注意不要打乱相应的原子顺序。
总结:提高SCF的计算精度(即提高力的计算精度至关重要)
转载请注明来源,欢迎对文章中的引用来源进行考证,欢迎指出任何有错误或不够清晰的表达。