92 +-------------------------- Warm Tips --------------------------+ Please Use These Features with CAUTION! +---------------------------------------------------------------+ ===================== 2D Materials Toolkit ====================== 921) Center Aomic-Layer along z direction 922) Resize Vacuum Thickness 923) Standardize 2D Crystal Cell 926) Elastic Constants for 2D Materials 927) Valence and Conduction Band Edges Referenced to Vacuum Level 929) Summary for Relaxed 2D Structure 0) Quit 9) Back ------------>> 923 -->> (1) Reading Structural Parameters from POSCAR File... -->> (2) Written POSCAR_NEW File!
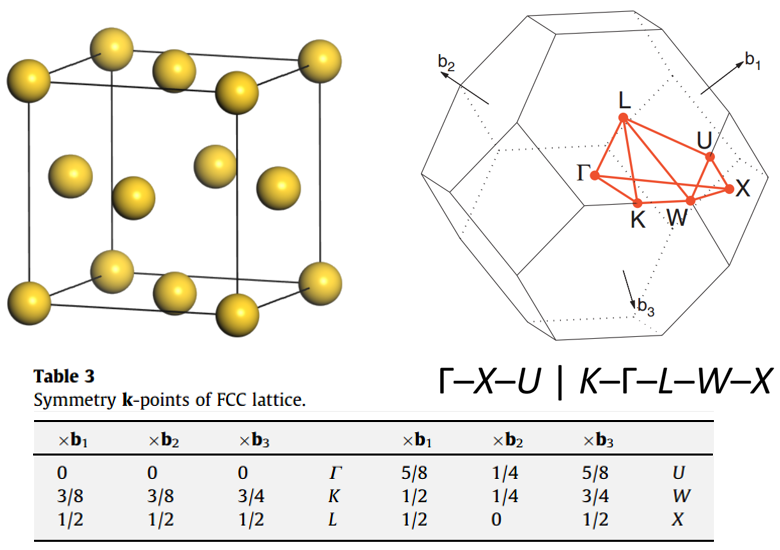
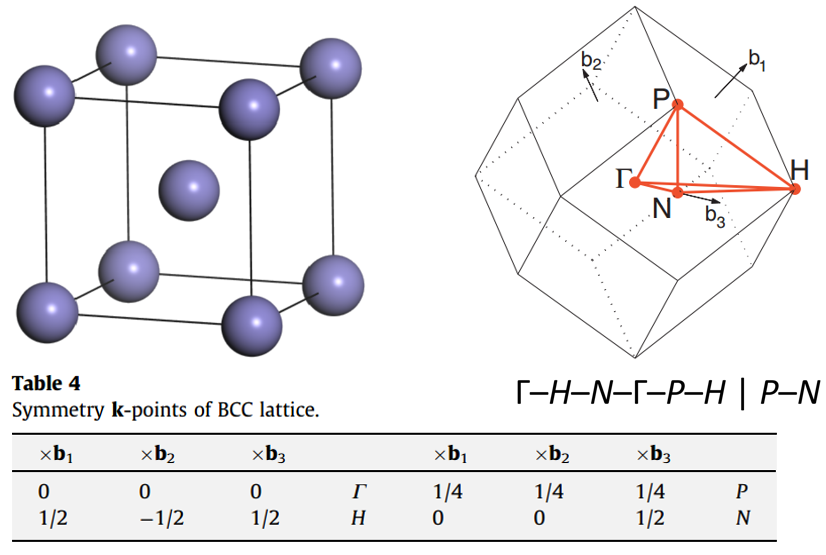
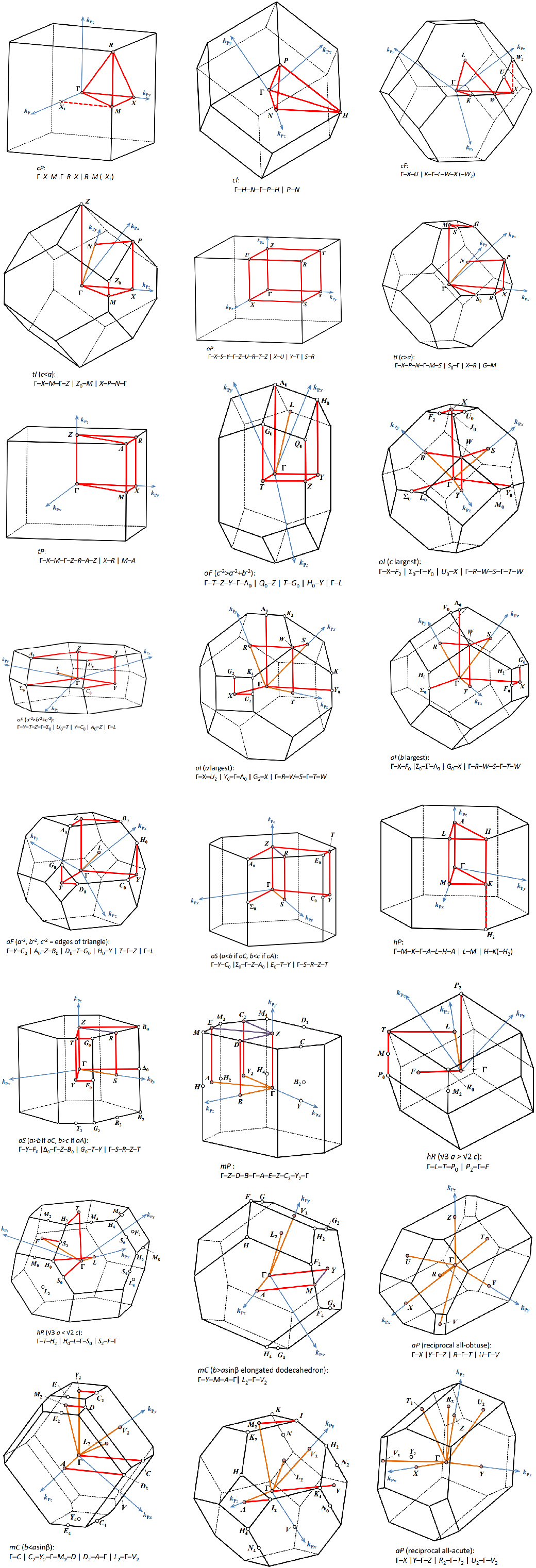
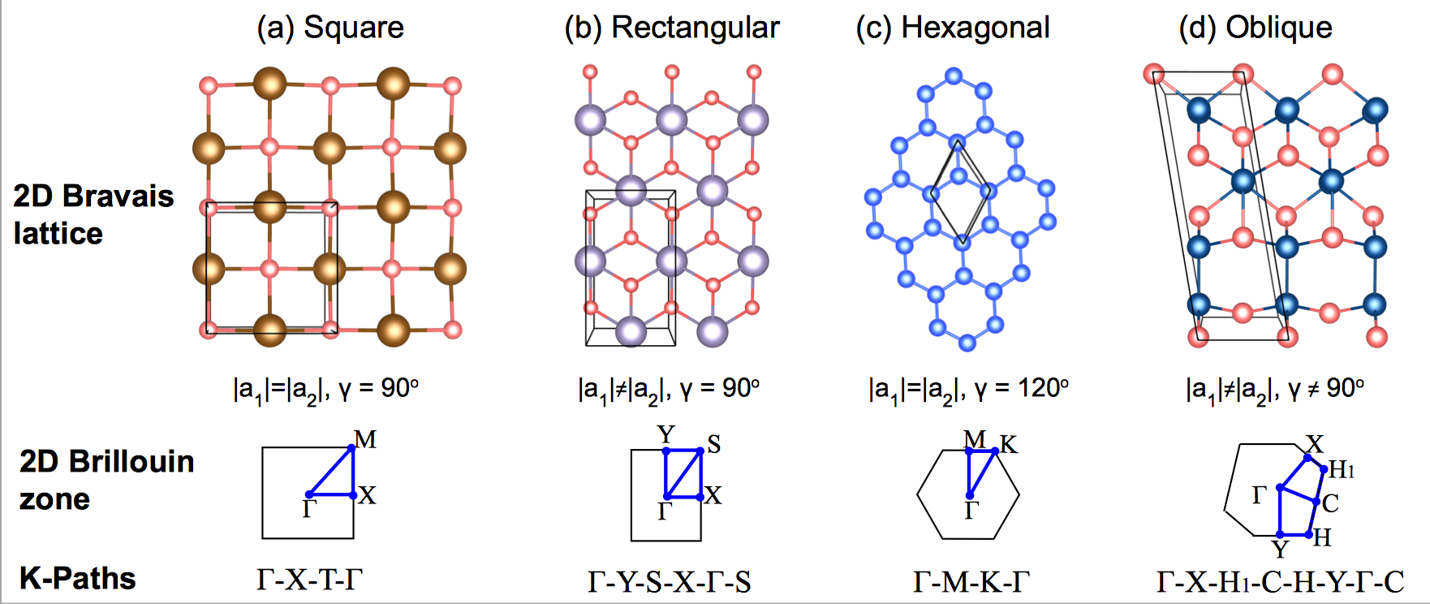
302 +-------------------------- Warm Tips --------------------------+ See An Example in VASPKIT/examples/seek_kpath/graphene_2D. This feature is still experimental & check the PRIMCELL.vasp file. +---------------------------------------------------------------+ -->> (1) Reading Structural Parameters from POSCAR File... +-------------------------- Summary ----------------------------+ The vacuum slab is supposed to be along c axis Prototype: AB2 Total Atoms in Input Cell: 3 Lattice Constants in Input Cell: 3.166 3.166 18.410 Lattice Angles in Input Cell: 90.000 90.000 120.000 Total Atoms in Primitive Cell: 3 Lattice Constants in Primitive Cell: 3.166 3.166 18.410 Lattice Angles in Primitive Cell: 90.000 90.000 120.000 2D Bravais Lattice: Hexagonal Space Group: 187 Point Group: 26 [ D3h ] International: P-6m2 Symmetry Operations: 12 Suggested K-Path: (shown in the next line) [ GAMMA-M-K-GAMMA ] +---------------------------------------------------------------+ -->> (2) Written PRIMCELL.vasp file. -->> (3) Written KPATH.in File for Band-Structure Calculation. -->> (4) Written HIGH_SYMMETRY_POINTS File for Reference.
High-symmetry points (in fractional coordinates). You can check them with the seekpath database [https://www.materialscloud.org/work/tools/seekpath]. 0.0000000000 0.0000000000 0.0000000000 GAMMA 0.3333333333 0.3333333333 0.0000000000 K 0.5000000000 0.0000000000 0.0000000000 M
If you use this module, please cite the following work: [1] V. Wang, N. Xu, J.-C. Liu, VASPKIT: A Pre- and Post-Processing Program for the VASP Code. http://VASPKIT.sourceforge.net. [2] V. Wang, Y.-Y. Liang, Y. Kawazeo, W.-T. Geng, High-Throughput Computational Screening of Two-Dimensional Semiconductors, arXiv:1806.04285.
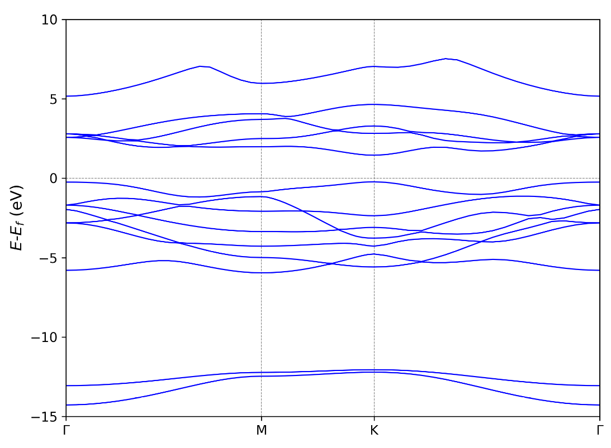
211 -->> (01) Reading Input Parameters From INCAR File... -->> (02) Reading Fermi-Energy from DOSCAR File... ooooooooo The Fermi Energy will be set to zero eV ooooooooooooooo -->> (03) Reading Energy-Levels From EIGENVAL File... -->> (04) Reading Structural Parameters from POSCAR File... -->> (05) Reading K-Paths From KPOINTS File... -->> (06) Written BAND.dat File! -->> (07) Written BAND_REFORMATTED.dat File! -->> (08) Written KLINES.dat File! -->> (09) Written KLABELS File! -->> (10) Written BAND_GAP File! If you want use the default setting, type 0, if modity type 1 0 /public1/home/pg2059/.local/lib/python2.7/site-packages/matplotlib/font_manager.py:1328: UserWarning: findfont: Font family [u'arial'] not found. Falling back to DejaVu Sans (prop.get_family(), self.defaultFamily[fontext])) -->> (11) Graph has been generated!
K-Label K-Coordinate in band-structure plots GAMMA 0.000 M 1.140 K 1.798 GAMMA 3.114 * Give the label for each high symmetry point in KPOINTS (KPATH.in) file. Otherwise, they will be identified as 'Undefined' in KLABELS file
BAND_GAP文件保存了VBM,CBM和带隙的数据:
1 2 3 4 5 6 7 8 9 10 11
+-------------------------- Summary ----------------------------+ Band Character: Direct Band Gap (eV): 1.6743 Eigenvalue of VBM (eV): -0.2257 Eigenvalue of CBM (eV): 1.4485 HOMO & LUMO Bands: 9 10 Location of VBM: 0.333333 0.333333 0.000000 Location of CBM: 0.333333 0.333333 0.000000 +---------------------------------------------------------------+
NOTE: The VBM and CBM are subtracted by the Fermi Energy.
如果安装有matplotlib的python环境,还能直接输出band.png能带图:
212,213,214功能绘制投影能带图:
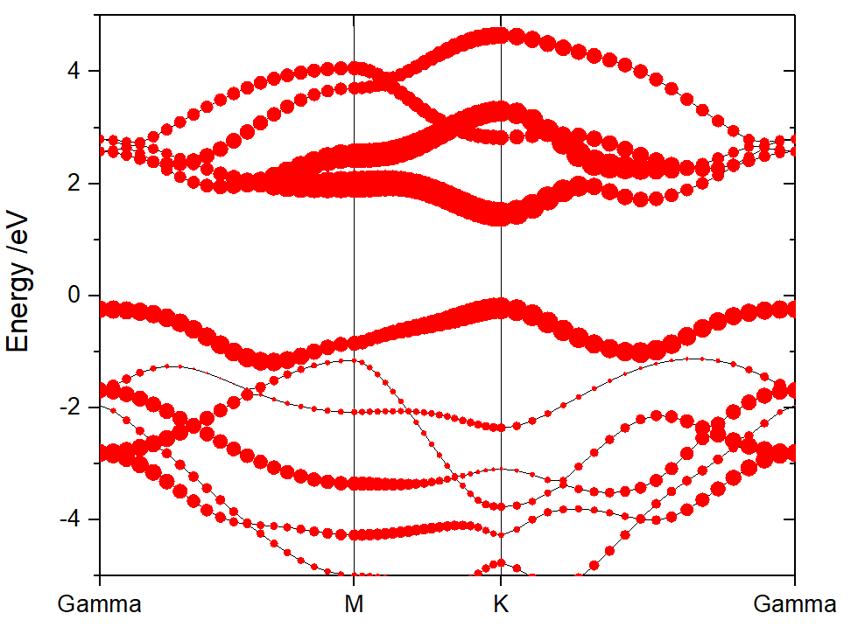
212) Projected Band-Structure for Selected Atoms,选择投影的原子,格式是自由格式的,可以同时选取几个原子或元素同时输出其投影能带数据,输入的方式可以是:
(1) 指定的原子序号,或序号集合:1-47824
(2) 指定的元素:CFeH
比如想要数据,1号和2号原子的投影能带数据,根据提示输入1-2:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
212 -->> (01) Reading Input Parameters From INCAR File... -->> (02) Reading Fermi-Energy from DOSCAR File... ooooooooo The Fermi Energy will be set to zero eV ooooooooooooooo -->> (03) Reading Structural Parameters from POSCAR File... -->> (04) Reading Energy-Levels From EIGENVAL File... -->> (05) Reading Band-Weights From PROCAR File... -->> (06) Reading K-Paths From KPOINTS File... |---------------------------------------------------------------| Input the element-symbol and/or atom-index to SUM [Total-atom: 3] (Free Format is OK, e.g., C Fe H 1-4 7 8 24) ------------>> 1-2 -->> (07) Written SELECTED_ATOM_LIST File! -->> (08) Written PBAND_A1.dat File! -->> (09) Written PBAND_A2.dat File! -->> (10) Written KLINES.dat File! -->> (11) Written KLABELS File!
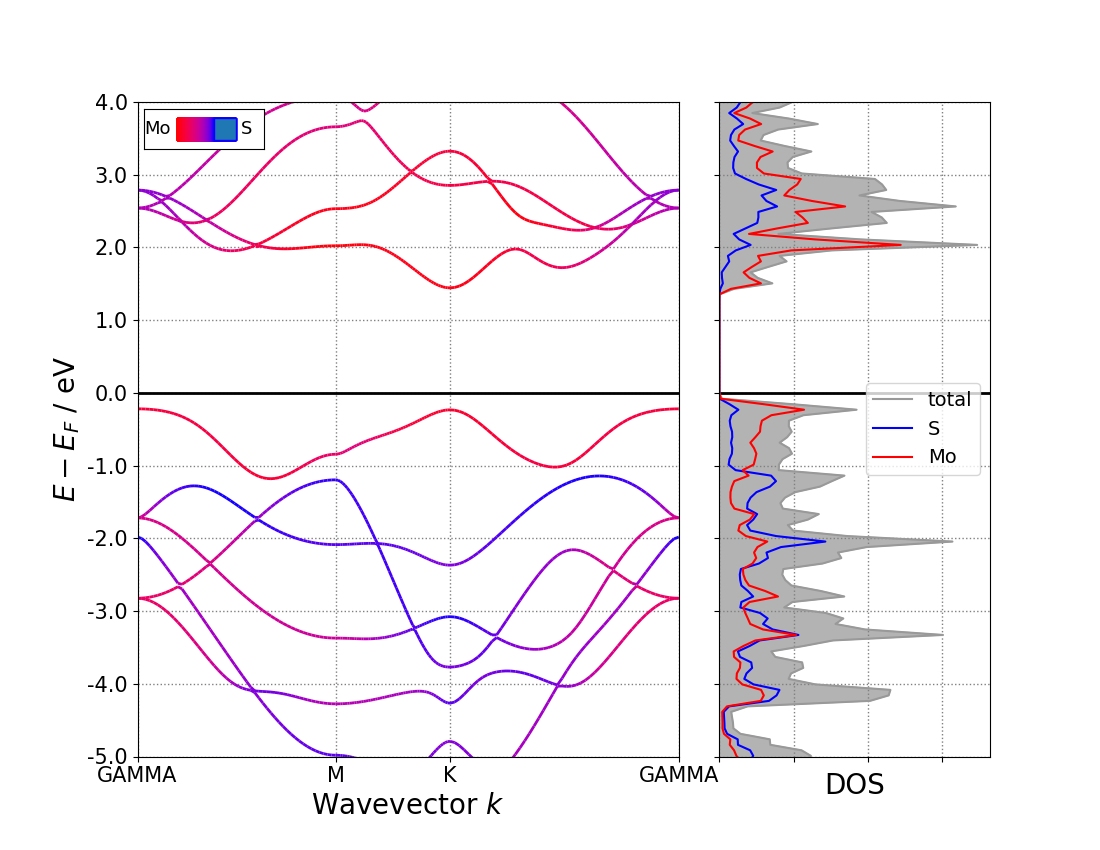
213) Projected Band-Structure for Each Element,投影到每种元素上:
1 2 3 4 5 6 7 8 9 10 11 12
213 -->> (1) Reading Input Parameters From INCAR File... -->> (2) Reading Fermi-Level From DOSCAR File... ooooooooo The Fermi Energy will be set to zero eV ooooooooooooooo -->> (3) Reading Structural Parameters from POSCAR File... -->> (4) Reading Energy-Levels From EIGENVAL File... -->> (5) Reading Band-Weights From PROCAR File... -->> (6) Reading K-Paths From KPOINTS File... -->> (7) Written PBAND_S.dat File! -->> (8) Written PBAND_Mo.dat File! -->> (9) Written KLINES.dat File! -->> (*) Written KLABELS File!
214) The Sum of Projected Band-Structure for Selected Atoms,该功能可以把几个原子或者元素的投影贡献加和:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
214 -->> (01) Reading Input Parameters From INCAR File... -->> (02) Reading Fermi-Energy from DOSCAR File... ooooooooo The Fermi Energy will be set to zero eV ooooooooooooooo -->> (03) Reading Structural Parameters from POSCAR File... -->> (04) Reading Energy-Levels From EIGENVAL File... -->> (05) Reading Band-Weights From PROCAR File... -->> (06) Reading K-Paths From KPOINTS File... |---------------------------------------------------------------| Input the element-symbol and/or atom-index to SUM [Total-atom: 3] (Free Format is OK, e.g., C Fe H 1-4 7 8 24)
------------>> Mo -->> (07) Written SELECTED_ATOM_LIST File! -->> (08) Written PBAND_SUM.dat File! -->> (09) Written KLINES.dat File! -->> (10) Written KLABELS File!