本文章为原创,版权归作者刘锦程所有,文章转载请先取得作者的同意,非常欢迎转发文章链接!严禁以任何方式挪用本文内容,用于以盈利为目的各种活动。
主讲人介绍:清华大学博士,长期从事表面催化和材料计算研究,对量化计算,第一性原理计算,分子动力学模拟有五年研究经验,精通 VASP,CP2K,Gaussian,ORCA熟练使用 JDFTx,Gromacs,ADF 等计算程序。近五年发表7篇一区一作文章。在表面催化,电催化,电池材料,过渡金属配合物等领域发表多篇文章。在JACS,Nat. Commun. ,ACS Catal.,Nano Energy, Natl. Sci. Rev. 等期刊上以第一作者发表纯理论计算文章,并与实验科学家合作在 Nat. Nanotech., Nat. Commun., PNAS,Adv. Mater., JACS 等期刊上发表多篇合作文章。
Google Scholar:
https://scholar.google.com/citations?user=hAkSR2wAAAAJ&hl=en
[课程报名请加企业微信]
1. 理论基础
第一性原理计算概述
第一性原理计算发展历史,历史引用,第一性原理计算的分类与应用。
计算研究思路原文解析例:
非铅双钙钛矿材料研究计算:Nature, 563, 541–545 (2018);
J. Phys. Chem. Lett. 2018, 9, 1, 43-48;
表面性质计算研究思路:
Nat. Catal.,2018, 1, 63;
Nature 2019, 565, 631-635.
Nat. Commun., 2018, 9, 1610.
Nature 2019, 565, 631-635
原文解析。
理论基础
量子力学概要,原子单位制,单位转化,狄拉克符号,算符,本征值(Eigenvalue)与本征函数(Eigenfunction),动量算符,动能算符,哈密顿算符,相对论效应,含时薛定谔方程,波函数的物理意义,概率密度,稳态薛定谔方程,Born-Oppenheimer (BO) 近似,变量分离,变分原理,HF(Hartree-Fock)法,单电子近似,Fork 算符 Fork 方程,求解 Hartree-Fork 方程,自洽场(self-consistent field, SCF)迭代,波函数的形式,基组,LCAO,HF方法的缺陷,交换相关,库伦相关,交换关联穴,相关穴,后HF方法。
密度泛函理论(DFT),Hohenberg-Kohn 定理,Kohn-Sham 理论,Kohn-Sham方程,交换相关泛函,LDA, GGA, meta-GGA, hybrid functional的形式和优缺点。自旋极化,Restricted、Unrestricted,晶体结构和固体理论,布拉维晶格( Bravais lattice),七种晶系,晶胞与晶体结构的表示,素晶胞,(primitive unit cell)和 惯用晶包 (conventional unit cell),32种点群,Schoenflies notation,Hermann–Mauguin notation,230种空间群,搜索晶体结构数据,CIF文件,ICSD,CCDC,COD,Materials Project,AMCSD,固体理论简介,布洛赫定理(Bloch’s theorem),傅里叶级数(Fourier series),周期性边界条件,波矢,平移不变性证明,倒易空间(Reciprocal space),布里渊区(Brillouin zone),K点,布里渊区对称性,高对称点,
第一性原理计算程序和学习方法
计算程序选择/介绍/特色/优缺点:
第一性原理计算程序:VASP,CP2K,Quantum-Espresso,SIESTA,CASTEP,Dmol3,ABINIT,wien2k,ADF-band,ATK,PWmat,JDFTx,等
量子化学程序:Gaussian,ORCA,ADF,NWChem,Molpro,Molcas,Turbomole,Q-chem,ADF,等
分子动力学模拟:Gromacs,Lammps,Amber,CHARMM,GULP,等
可视化和建模程序选择:MS,Vesta,Jmol,Gaussview,VMD。
后处理程序选择介绍:VASPKIT,qvasp,p4vasp
第一性原理书籍推荐。第一性原理学习资料。第一性原理计算学习资料链接大全
2. 超算服务器的使用实战
•Linux权限的概念
•Linux基本命令
•Linux系统特殊字符
•vi的使用基础
•Linux建议的文件目录管理方法
•队列系统的使用(pbs,lsf,slurm,天河改的slurm)
•如何在超算上编译VASP
如何在自己的机器上编译VASP
如何编译更多功能VASP(VTST, vaspsol, 选择性优化晶胞矢量, ssNBO, ssAdNDP)
3. VASP的输入文件
INCAR,KPOINT,POTCAR,POSCAR,这四个文件的详细解读,和操作方法,包括: INCAR,KPOINTS 文件的生成方法,选取K点的原则,自动生成k点方法,POTCAR 的生成方法,不同赝势文件的选取,赝势方法的基本原理。POSCAR 结构文件的生成方法,分数坐标,笛卡尔坐标。
4. VASP的输出文件
VASP 输出文件:输出文件的分析,OUTCAR,OSIZCAR,stdout,CHGCAR 文件详解,其他输出文件的详细解读。
5. 建模
模块介绍,界面和功能,建模模块的功能和使用,切面加真空层,建立带根号的表面模型 (旋转晶格矢量)。
- 实例一,构建 Na(111)/graphene 异质结,
- 实例二,文献重复:构建 g-C3N4/TiO2 的界面材料,
- 实例三,gamma-Al2O3(010) 模型的构建,
- 实例四,alpha-Al2O3(0001) 面的构建和悬挂基团的选择,
- 实例五,异质结建模方法,
- 实例六,GaN/MoS2异质结且半导体表面用赝氢饱和的建模方法,
- 实例七,做满足文章 TOC 要求的图。
6. 收敛性测试
7. INCAR 几十种常见关键词全解析与建议使用表
8. 晶胞优化
结构优化的技巧和注意事项。选择性优化晶胞矢量。不同泛函的使用。原子电荷计算分析。实例一:theta-Al2O3 单点能计算和结构优化,实例二:文献重复不同泛函比较。
带电荷的体系计算.
9. 态密度, 10. 能带计算
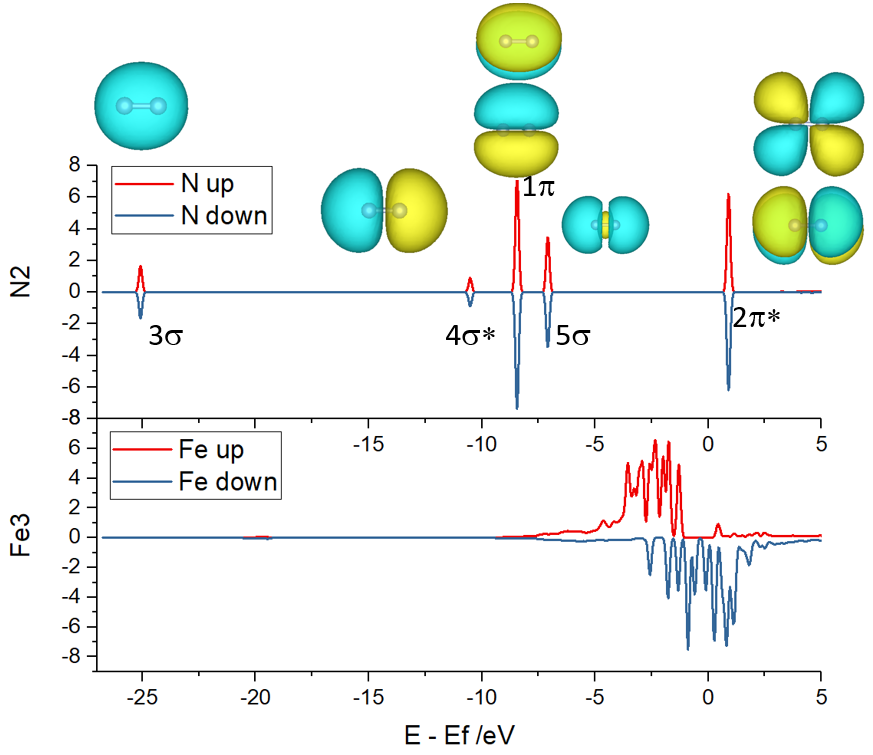
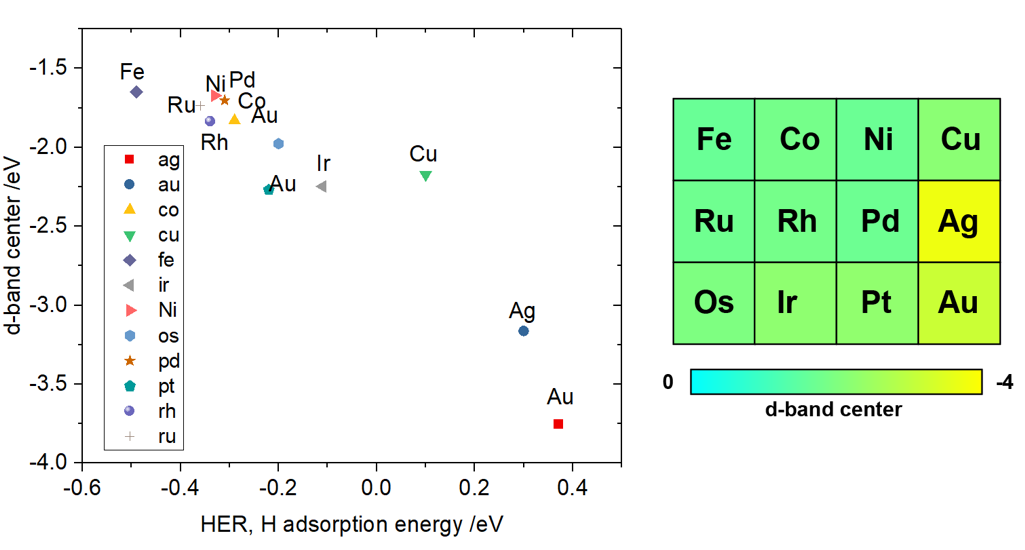
态密度和能带形成的基本理论,案例分析(单原子催化剂,表面离子缺陷态,表面态,d带中心,催化吸附的轨道相互作用模型),态密度计算方法,p4vasp和VASPKIT后处理分析,态密度的积分,pymatgen自动画态密度图,K点选取和ISMEAR对态密度计算影响,DOSCAR详解,过渡金属金属的d带中心计算和催化吸附的关系计算,N2分子和表面团簇催化剂的轨道相互作用模型分析。
轨道相互作用基础知识,高斯计算提取分子轨道,载体和吸附分子态密度作用分析,CO/Ni,N2/Fe3催化剂的轨道相互作用模型分析。bilibili视频
实例一:Theta-Al2O3 的态密度,
实例二:Ir,Pt,Au 的 d 带中心计算,
实例三:顶刊重复 N2/Fe3 Al2O3 DOS 计算。
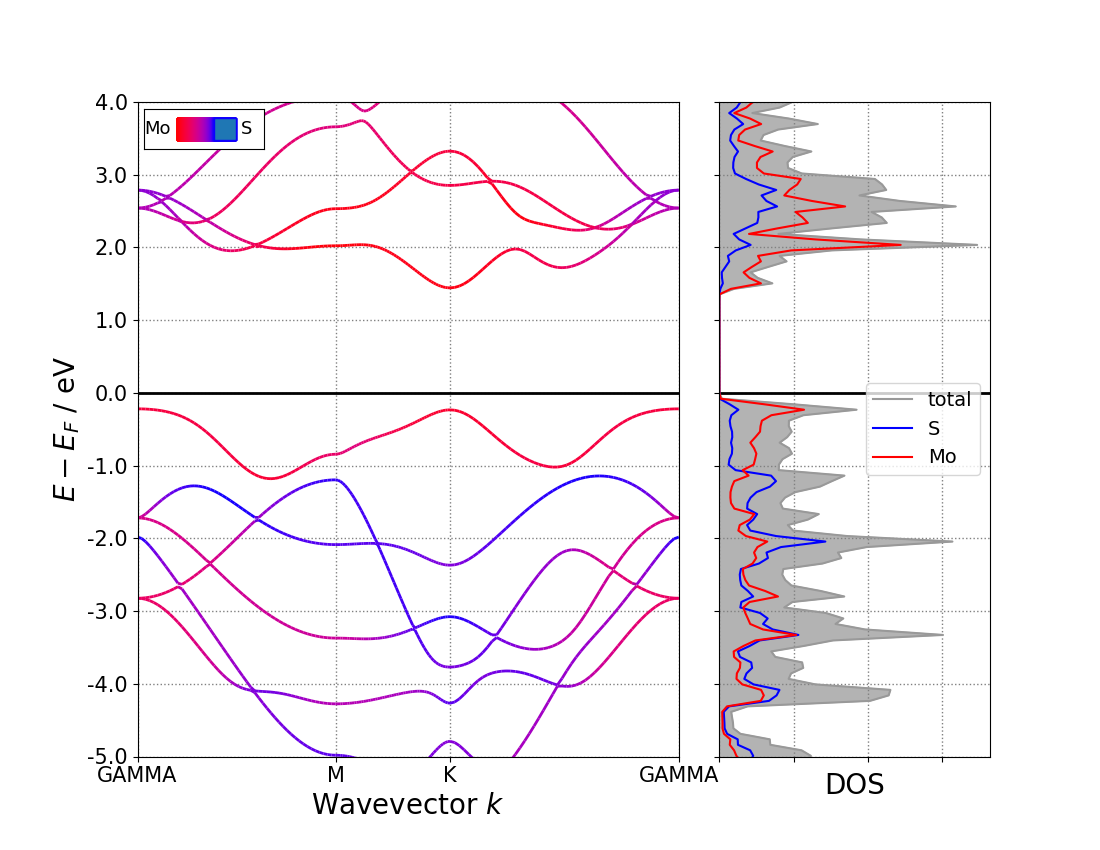
能带计算:生成布里渊区中K点路径的方法,选取路径的技巧,VASPKIT 自动生成k点路径,see kpath 的使用。能带结构的后处理,p4vasp-origin 处理能带图,pymatgen 生成能带图,VASPKIT 生成能带图。
实例一:金属 Au 能带结构,
实例二:theta-Al2O3 的能带结构,
实例三:二维材料 不同层数 MoS2 的能带结构,
杂化泛函 HSE 对能带结构的影响,
实例四:对比 HSE 和 PBE 的能带结构。
11. 解决VASP收敛和报错问题
12. 计算的艺术——平衡速度与精度
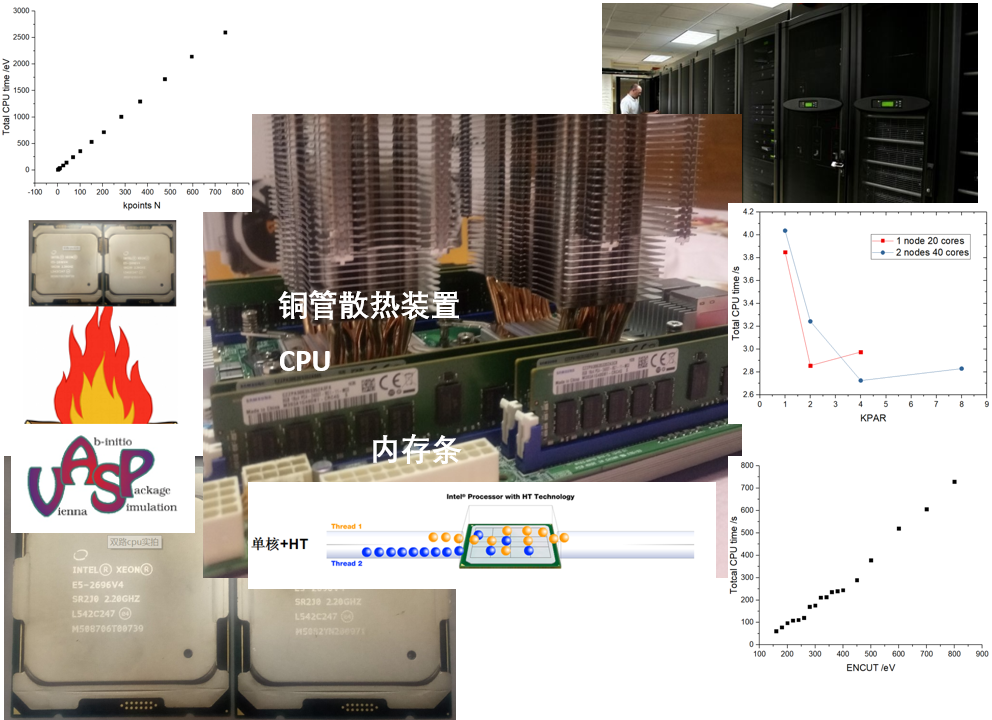
13.表面物理化学基础知识,14. 表面能
原子堆积方式,FCC、HCP,Miller指数定义,高miller指数晶面与台阶面,表面弛豫与重构,对称和非对称的slab模型,对称和非对称表面的表面能计算,金属表面能和功函数据库,原子坐标的固定,纳米颗粒的结构和表面能关系,Wulff construction计算,纳米催化中的晶面效应。
15. 静电势和功函
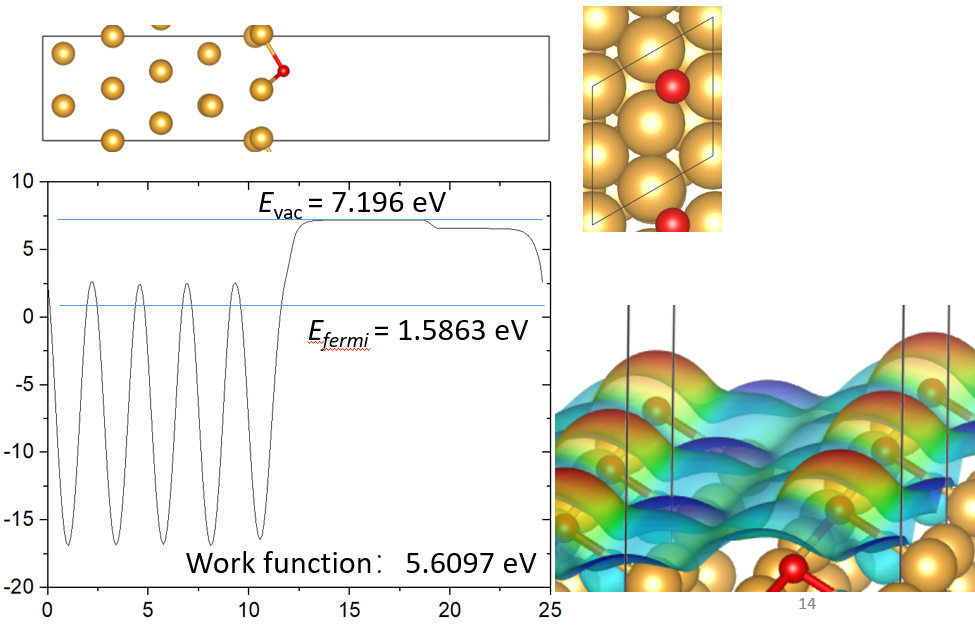
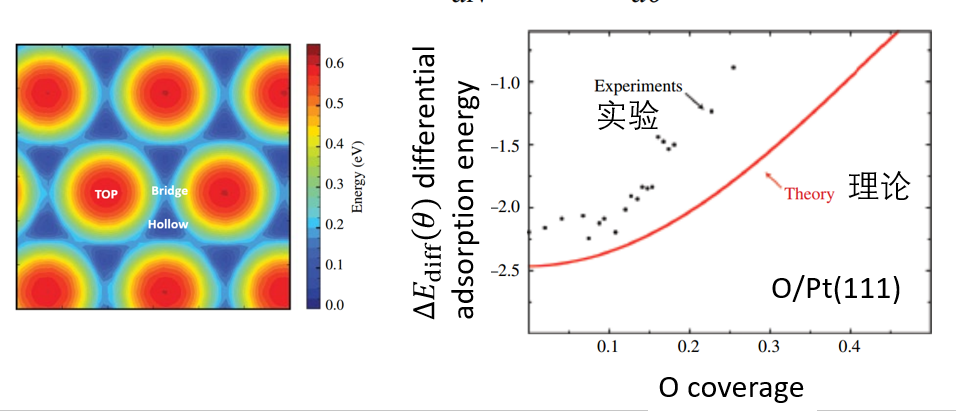
16. 表面吸附
吸附类型(化学吸附、物理吸附、解离吸附),吸附能计算,吸附位点,吸附计算注意事项,吸附物种间的相互作用,覆盖度,平均吸附能,最后一分子的吸附能。
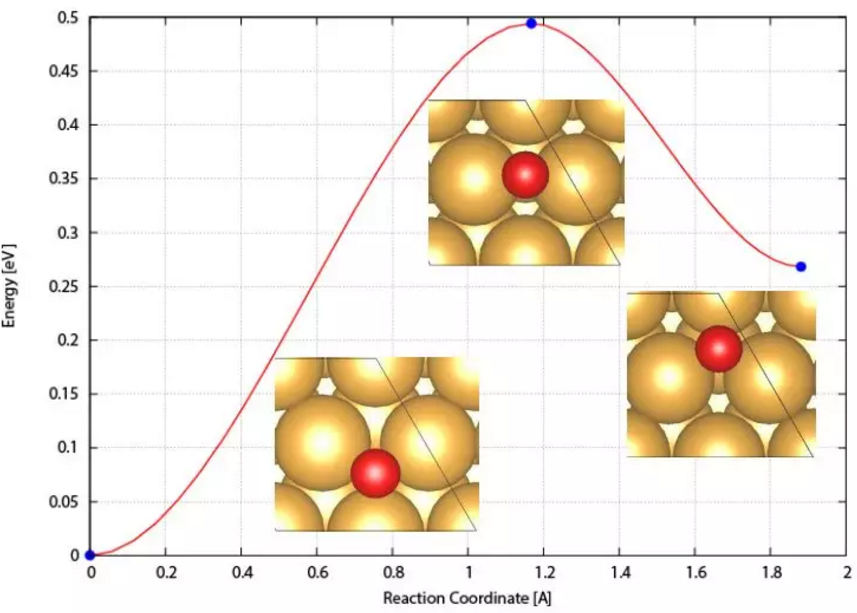
17. 过渡态搜索
过渡态判据,无过渡态反应类型,寻找过渡态方法综述,chain-of-states搜索方法和原理,传统PEB方法(plain elastic band)方法遇到的问题,NEB中nudge过程原理,Climbing Image原理,判断插点个数,插点方法,nebmovie.pl源码修改,生成movie判读插点合理性,CINEB的INCAR关键词和计算方法,VTST优化算法解析,nebresults.pl,nebbarrier.pl, nebspline.pl, nebef.pl等VTST脚本,neb.dat、spline.dat、exts.dat、mep.eps文件自动判断NEB路径上的极值点,Dimer方法原理,选择Dimer,平移Dimer过程,Dimer发展历史,Dimer的INCAR关键词和计算方法,MODECAR生成,DIMCAR、CENTCAR和NEWMODECAR解读,生成DIMER方向movie预判过渡态计算是否成功,VTST过渡态计算收敛判断,过渡态搜索常见问题和解决方案。扩散系数与离子电导计算。
18. 频率计算
Hessian矩阵,力常数矩阵对角化,孤立和吸附分子振动区别,零点振动能,有限位移和DFPT方法频率计算INCAR,Jmol观察振动,振动数据提取和分析。晶体弹性常数/弹性模量计算.
19. 热力学数据计算与电催化
系综配分函数,分子配分函数,热力学量与配分函数的关系,内能、亥姆霍兹自由能、焓、吉布斯自由能、熵、等容热容、等压热容,理想气体假设,能量成分的拆分,平动、转动、振动、电子跃迁的配分函数,和其对各种热力学量的贡献计算,振动频率、温度和熵的关系,0K热力学数据校正计算,实际温度热力学数据校正计算,VASPKIT计算吸附分子热力学量,吸附分子和孤立分子的热力学量计算区别。气体分子的自由能校正,查实验数据热力学表JANAF-NIST,使用Gaussian程序做频率计算得到热力学数据,VASPKIT计算孤立分子热力学量。
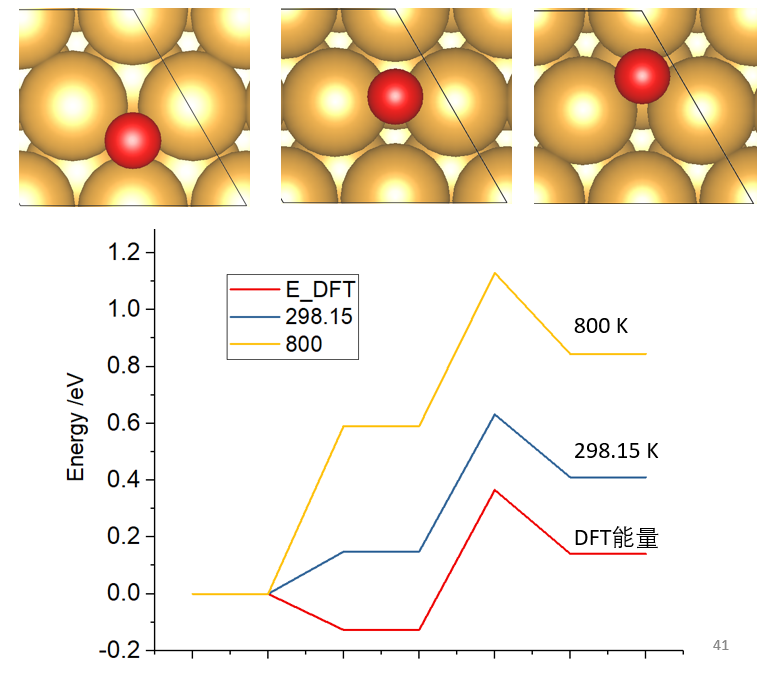
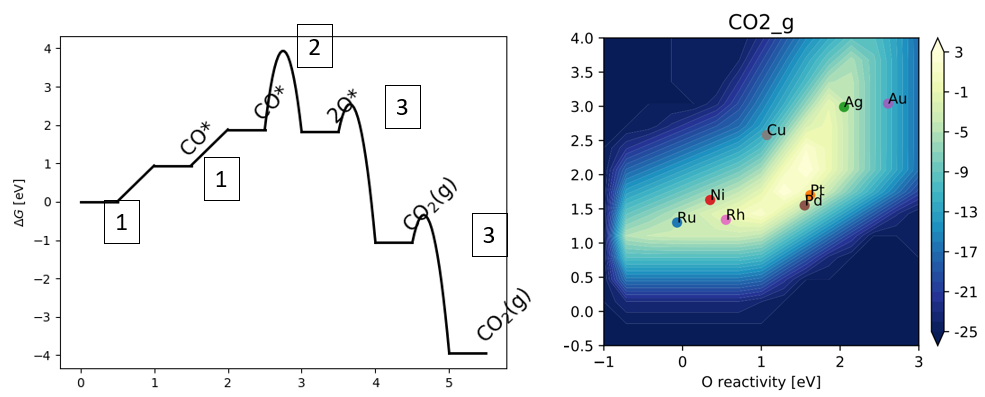
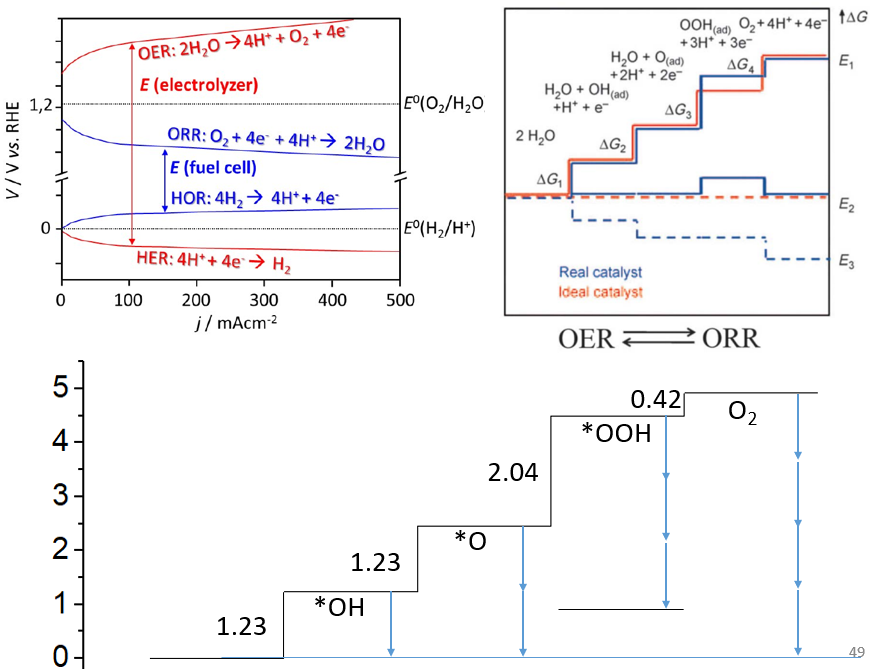
20. 磁性体系计算
自旋量子数,自选多重度,自旋角动量,第一性原理计算中 电荷的设定,电子所固有的自旋;电子绕核旋转的轨道角动量;外加磁场感生的轨道磁矩改变。铁磁性(ferromagnetic)反铁磁性(antiferromagnetic)亚铁磁性(ferrimagnetic)顺磁性(Paramagnetic)。
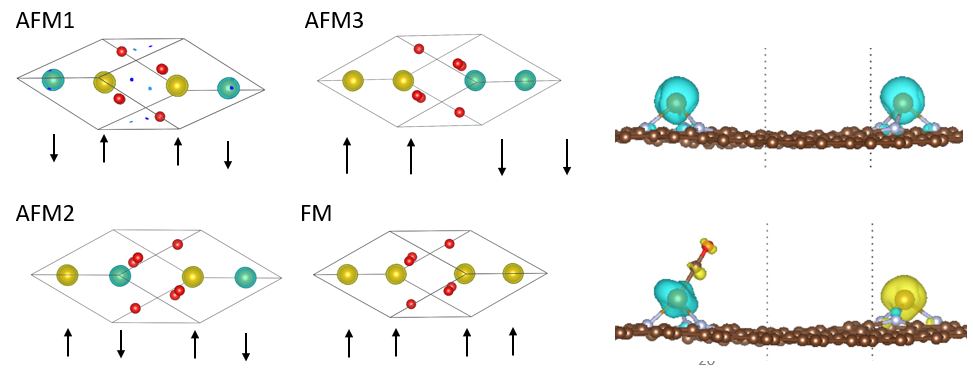
21. DFT + U 简介,复杂体系参数设置
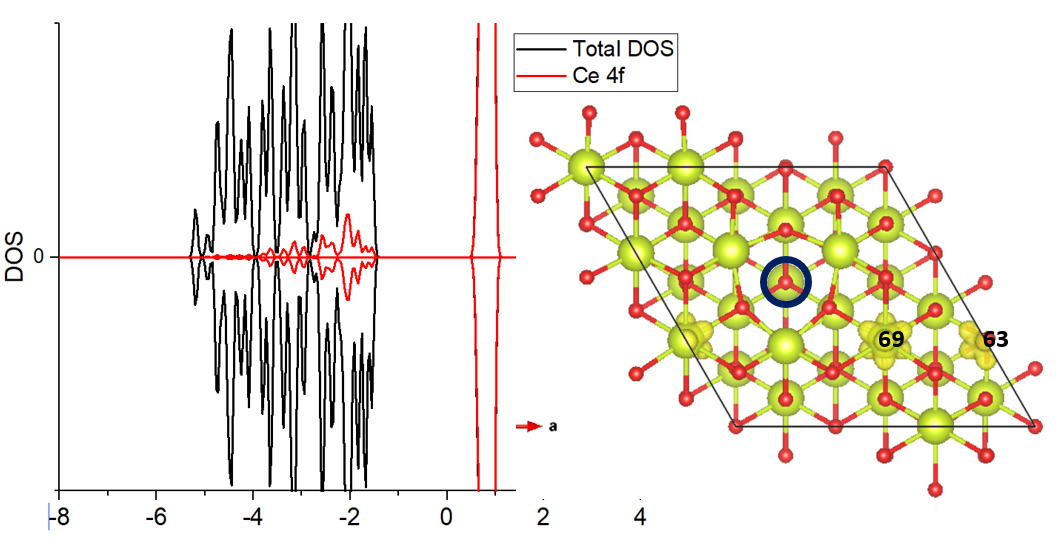
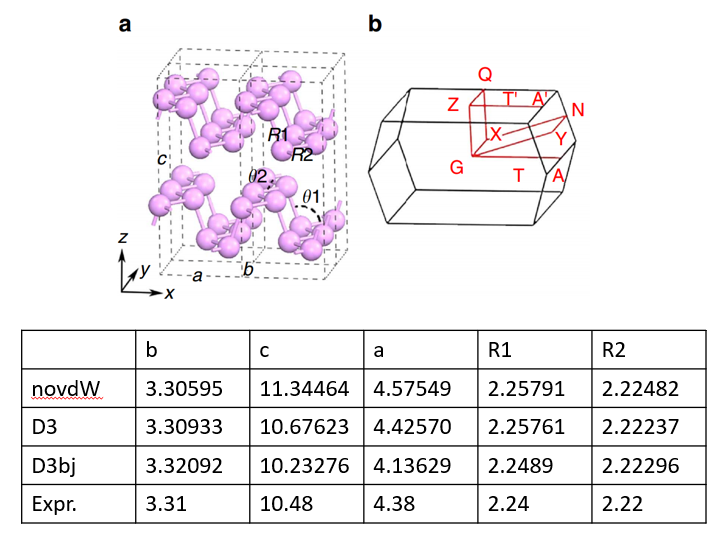
22. 范德华校正简介
23. 电子结构分析
常见VASP第一性原理计算中的电子结构分析手段总述。
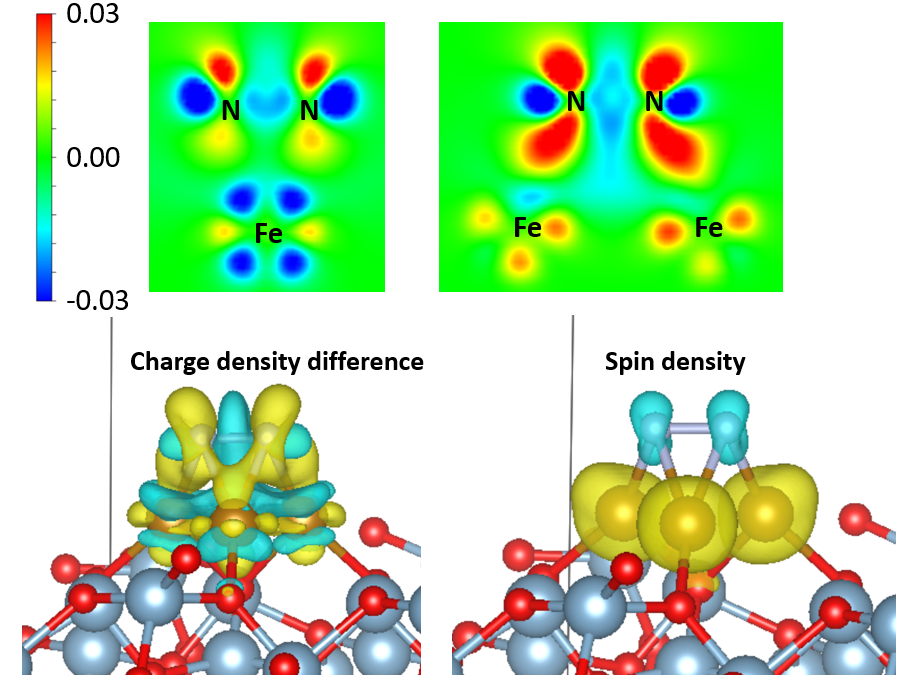
电荷密度和原子电荷,CHCGAR的格式,分解CHGCAR成tot和mag,VESTA作等值面图,二维切面图。VASPKIT处理电荷密度文件,电荷密度差分,吸附分子CO,N2吸附前后的电荷密度差,石墨烯插锂前后的电荷密度差;变形电荷密度;外场下的电荷密度差;平面平均的电荷密度差。Partial charge density计算方法流程,实空间波函数提取,绘制分子的分子轨道图,绘制VBM和CBM的电荷密度/波函数,绘制缺陷态/隙间态的电荷密度/波函数,研究激发电子的分布,STM和Partial Charge Density对应关系。Bader电荷,全电子电荷密度生成,AIM(atom-in-molecule)电荷划分方法,按照原子电荷着色的结构图。
转载请注明来源,欢迎对文章中的引用来源进行考证,欢迎指出任何有错误或不够清晰的表达。