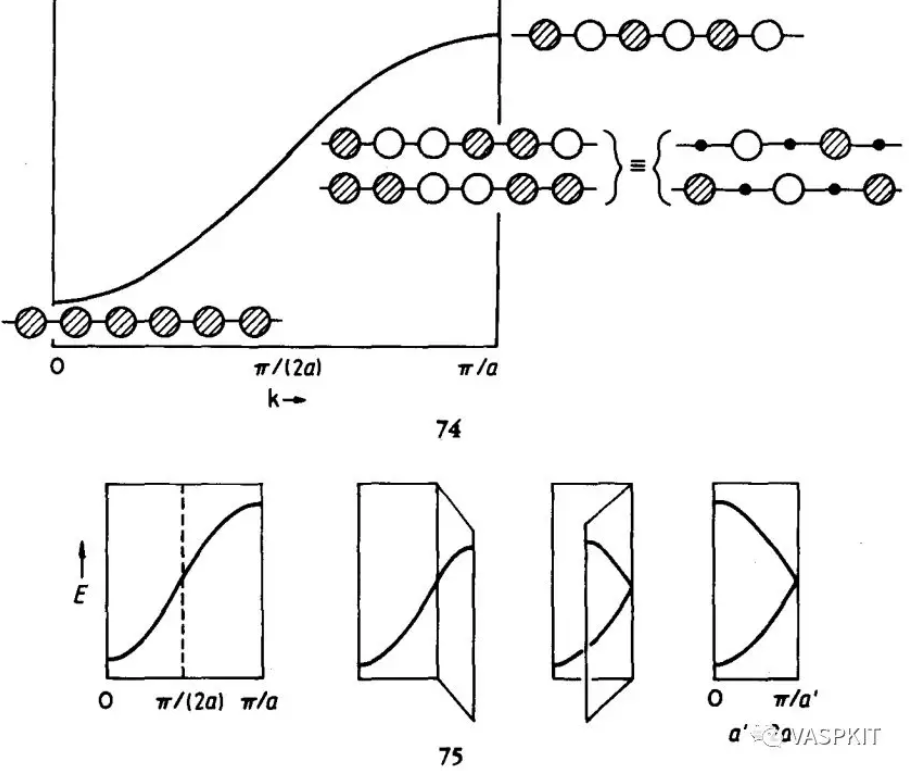
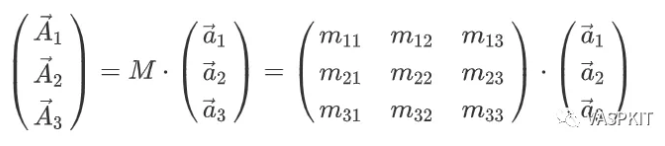Read transformation matrix from the TRANSMAT.in file if it exists. 第一行是注释行 2 0 0 # must be three integers 0 3 0 # must be three integers 0 0 3 # must be three integers
VASPKIT -task 400 +---------------------------------------------------------------+ | VASPKIT Version: 1.00.RC (17 Jun. 2019) | | A Pre- and Post-Processing Program for VASP Code | | Running VASPKIT Under Command-Line Mode | +---------------------------------------------------------------+ -->> (01) Reading Structural Parameters from POSCAR File... Enter the new lattice verctor a in terms of old: (MUST be three integers, e.g., 1 2 3) 4 0 0 Enter the new lattice verctor b in terms of old: 2 4 0 Enter the new lattice verctor c in terms of old: 0 0 1 +-------------------------- Summary ----------------------------+ The Transformation Matrix P is: 4 0 0 2 4 0 0 0 1 Lattice Constants in New Cell: 12.762 11.052 17.659 Lattice Angles in New Cell: 90.00 90.00 90.00 Total Atoms in New Cell: 48 Volume of New Cell is 16 times of the Old Cell +---------------------------------------------------------------+ -->> (02) Written SUPERCELL.vasp File
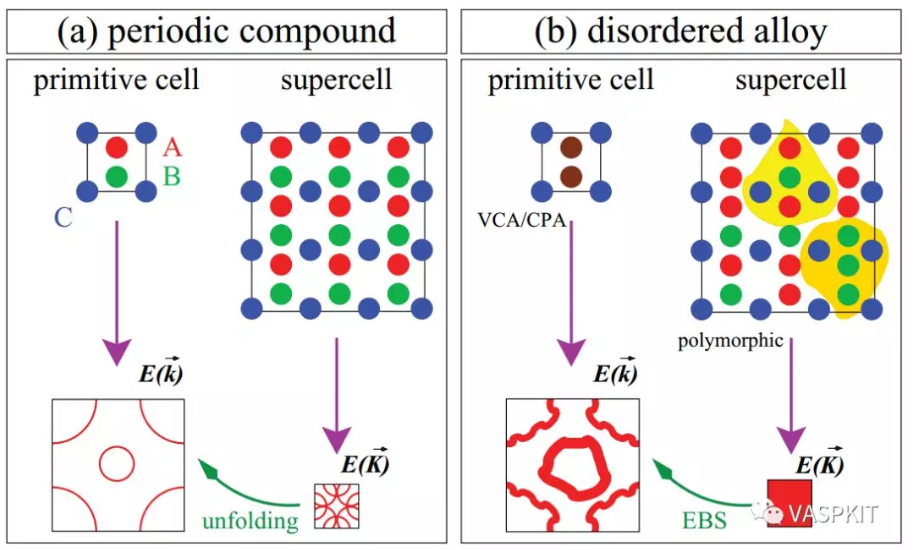
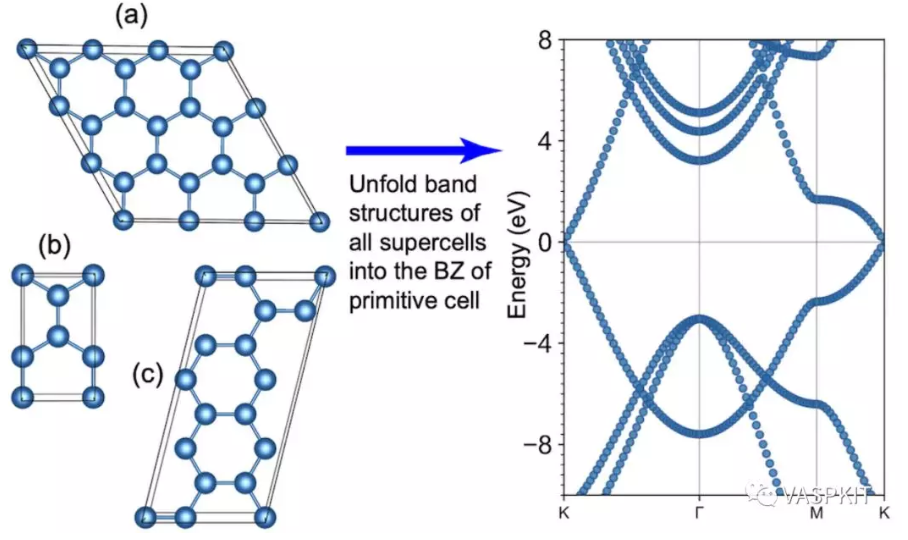
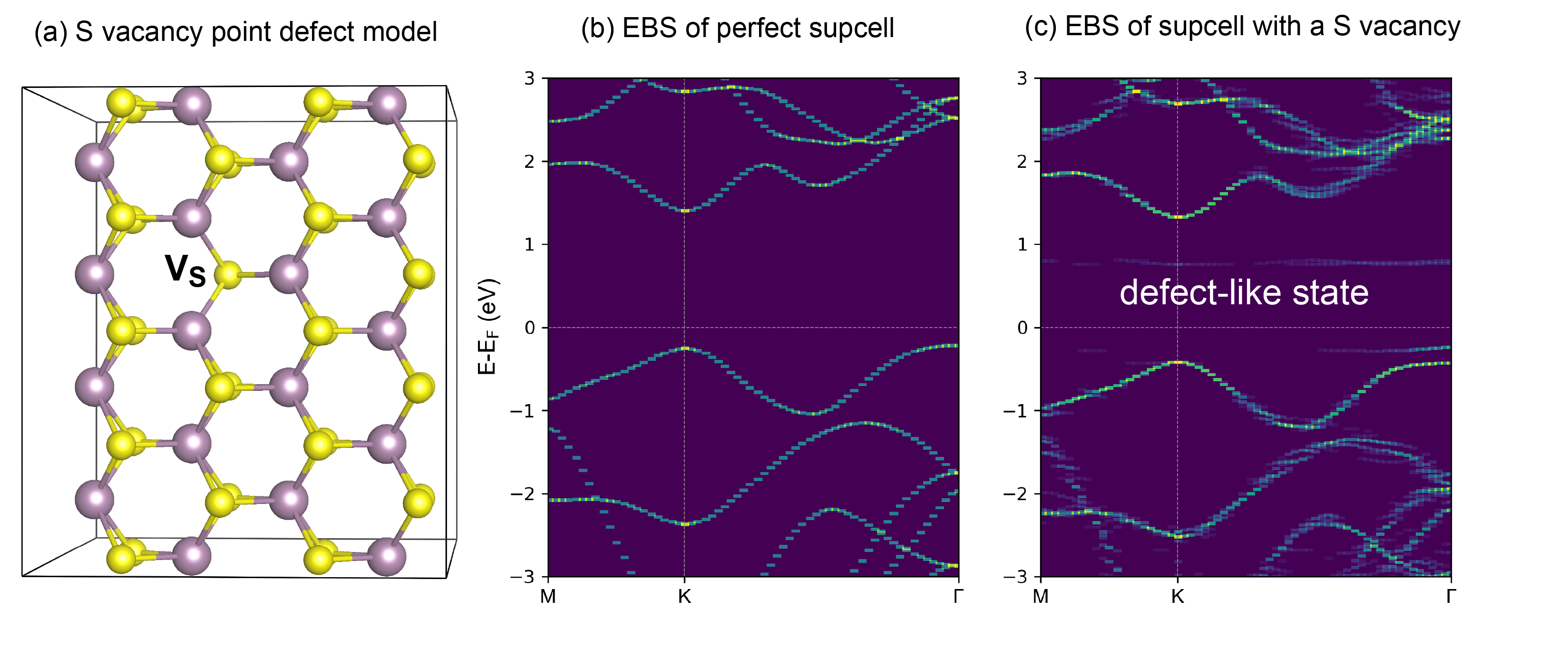
------------>> 28 +-------------------------- Warm Tips --------------------------+ See some examples in VASPKIT/examples/band_unfolding. Only Support KPOINTS file Generated by VASPKIT. Please Set LWAVE= .TRUE. in the INCAR file. +---------------------------------------------------------------+ ================== Band-Unfolding ============================== 281) Generate KPOINTS File for Band-Unfolding Calculation 282) Calculate Effective Band Structure
0) Quit 9) Back ------------->> 2 +-------------------------- Warm Tips --------------------------+ Input Resolution Value to Determine K-Mesh for SCF Calculation: (Typical Value: 0.02-0.03 is Generally Precise Enough) ------------>> 0.03 Input Resolution Value along K-Path for Band Calculation: (Typical Value: 0.02-0.04 for DFT and 0.04-0.06 for hybrid DFT) ------------>> 0.03 +---------------------------------------------------------------+ -->> (02) Readin Transformation Matrix from TRANSMAT.in file... -->> (03) Reading K-Paths From KPATH.in File... +-------------------------- Summary ----------------------------+ K-Mesh for SCF Calculation: 3 3 1 # 注意这部分根据超胞的倒格矢长度产生 The Number of K-Point along K-Path 1: 21 # 注意这部分根据原胞的倒格矢长度产生 The Number of K-Point along K-Path 2: 43 # 注意这部分根据原胞的倒格矢长度产生 +---------------------------------------------------------------+
------------>> 282 +-------------------------- Warm Tips --------------------------+ Current Version Only Support the Stardard Version of VASP code. +---------------------------------------------------------------+ -->> (01) Reading the header information in WAVECAR file... +--------------------- WAVECAR Header --------------------------+ SPIN = 1 NKPTS = 68 NBANDS = 252 ENCUT = 280.00 Coefficients Precision: Complex*8 Maximum number of G values: GX = 18, GY = 16, GZ = 25 Estimated maximum number of plane-waves: 30159 +---------------------------------------------------------------+ -->> (02) Start to read WAVECAR file, take your time ^.^ Percentage complete: 25.0% Percentage complete: 50.0% Percentage complete: 75.0% Percentage complete: 100.0% -->> (03) Readin Transformation Matrix from TRANSMAT.in file... -->> (04) Reading Fermi-Energy from DOSCAR File... ooooooooo The Fermi Energy will be set to zero eV ooooooooooooooo -->> (05) Reading KPOINTS_MAPPING_TABLE.in file... -->> (06) Reading K-Paths From KPATH.in File... -->> (07) Start to Calculate Effective Band Structure... Percentage complete: 25.0% Percentage complete: 50.0% Percentage complete: 75.0% Percentage complete: 100.0% -->> (08) Written EBS.dat File! -->> (09) Written KLABELS File!