本文章为原创,版权归作者刘锦程所有,文章转载请先取得作者的同意,非常欢迎转发文章链接!严禁以任何方式挪用本文内容,用于以盈利为目的各种活动。
相关内容:基于VASP的Periodic NBO编译和简单测试
Periodic NBO方法的局限性:Periodic NBO程序只能处理双中心键,分子体系的NBO程序最多只能处理3中心键。
对于3中心以上键,比如离域大p键,NBO就很难处理了,虽然借助于自然共振理论(NRT),NBO可以对苯环给出合理的共振模型,但是其对多中心键没有办法描述。
AdNDP是Boldyrev课题组在2008年发表的。在NBO的思路基础上可以继续搜索3c-2e、4c-2e、5c-2e、6c-2e。具体的原理可以阅读原文,
Phys. Chem. Chem. Phys., 2008, 10, 5207–5217
虽然这个方法非常的新,但是迅速得到了广泛的应用,已经被引用500多次,不乏一些顶尖期刊引用。
因为一系列有意思的分子和材料存在离域的电子结构,AdNDP可以把这种离域的电子结构以多中心键的方式呈现出来。
AdNDP 计算流程:
原子块对角化 + OWSO正交化得到自然原子轨道为基的密度矩阵P(NAO),和NBO的处理方法完全一样。
搜索单中心键,需要设置占据数阈值
搜索2c-2e键,需要设置占据数阈值
搜索3c-2e键,需要设置占据数阈值
依次搜索4c-2e键,5c-2e键,6c-2e键
注:每一步的搜索结果都会影响后续的搜索,对选中的多中心键,对应的
P
(NAO)
里的成分剔除。
AdNDP 和 ssAdNDP
分子体系计算AdNDP最好用的程序是Multiwfn,Boldyrev组也开发了名为AdNDP的程序,但非常难用,不如Multiwfn灵活。
周期性体系,计算AdNDP的程序只有Alexander I. Boldyrev和J. R. Schmidt(periodic NBO作者)共同开发的名为ssAdNDP的程序可以做。
http://ion.chem.usu.edu/~boldyrev/ssadndp.php
这个程序年久失修,非常难用,搜索轨道时候往往要不断的重启任务,虽然方法很好,目前还没有更好的程序可以做。
Physical Chemistry Chemical Physics, 2013, 15(14): 5022-5029
ssAdNDP 编译
参考 periodic NBO的编译方法,用intel2013编译器。
第一步:重新编译VASP,和periodic NBO的方法一样。
第二步:编译projection.exe,方法和编译periodic NBO的projection.exe基本类似。
只需要修改makefile里
MKLROOT=/opt/intel/composer_xe_2013_sp1.2.144/mkl/
第三步:编译ssadndp.exe,用mkmf生产Makefile 。
修改Makefile.template里的intel mkl路径
chmod +x mkmf
./mkmf -t Makefile.template -p ssadndp.exe
make
编译成功以后生成ssadndp.exe
第四步:编译visual.exe
ifort visual.f90 -o visual.exe
ssAdNDP 应用实例
2D Borphene, MgB2, Na8BaSn6 (Phys. Chem. Chem. Phys., 2008, 10, 5207–5217)
FeB2 (Nano Lett. 2016, 16, 6124−6129)
FeB6 (J. Am. Chem. Soc. 2016, 138, 5644−5651)
Be5C2 (Nat. Commun. 2016, 7, 11488 2016)
Cu2Si (J. Am. Chem. Soc. 2015, 137, 2757−2762)
ssAdNDP 的搜索技巧
- 从低中心数的键开始,逐渐增加中心数搜索。不可直接从多中心的键开始。
- 搜索到的AdNDP轨道不是全都选择,可以选择性的挑出部分符合化学直觉的轨道,一般来说挑出的轨道占据数都要尽可能大,且最终残留的电子数最少。
- AdNDP搜索有很强的人为因素!同一个体系有多种轨道挑选的方案,可以人为的选出合理的轨道。所以,不同的人做出来的多中心键的组合方式有所差异是非常正常的。
- 只要AdNDP的结果能够对体系的离域化学键特征做出合理的解释就成功了。AdNDP没有唯一解。
测试:
原文中MgB2 多中心键研究。(Physical Chemistry Chemical Physics, 2013, 15(14): 5022-5029)
作为一个范例, Boldyrev在ssAdNDP的原文中研究了经典超导材料MgB2中可能的多中心键。
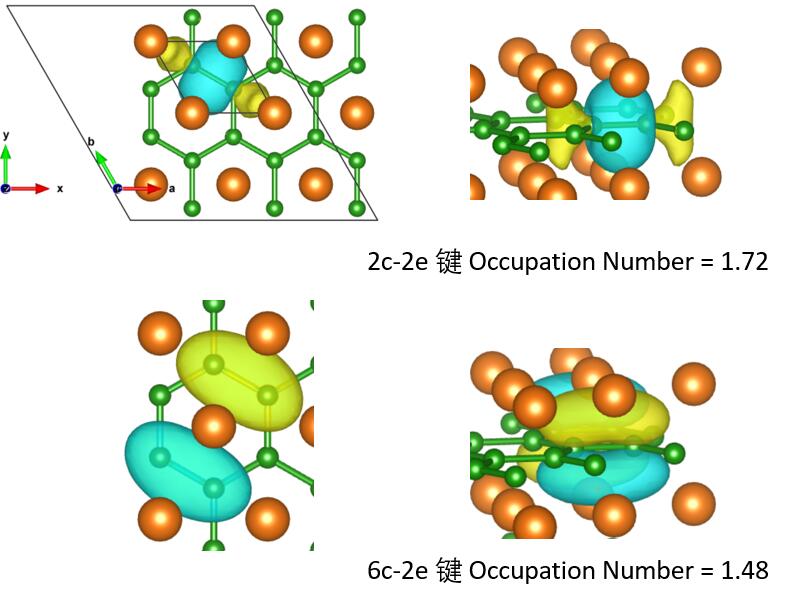
MgB2晶胞中有2个B原子和一个Mg原子。价电子数分别为3和2。则体系中共有8个电子。
作者搜索出来5个多中心键,是因为轨道占据数较低,更够把8个电子分配到5个轨道里,这种做法我并不建议。
第一步:在materials project网站搜索MgB2结构,mp-763
第二步:VASP优化这个结构。
第三步:计算MgB2单点,得到后续projection计算需要的wavefunction.dat文件。注意计算完成之后要把CONTCAR里的第六行删掉,因为此程序还是用的VASP4的格式。
VASP计算的注意事项和 periodic NBO完全一样。
第四步:准备基组文件
在EMSL上准备basis.inp文件,Mg和B的基组用6-31g*
第五步:运行./projection.exe basis.inp wavefunction.dat NBO.out,将平面波基组投影到高斯基组,会生成NBO.out,NBO_mat.out,band_spillover.out,spillover.out文件。
注意:这里运行的./projection.exe 是和SSAdNDP配套的投影程序,我尝试用periodic NBO配套的projection.exe 并没有成功。
第六步:准备SSAdNDP.inp文件,计算SSAdNDP
运行./ssadndp.exe SSAdNDP.inp
该文件是计算SSAdNDP的输入文件,参数控制都在这个文件里设置,这个文件的参数设置非常不灵活。详细内容可以参阅手册。
由于AdNDP不能全自动完成,所以要边搜索边手动挑选出合理的轨道,不断的保存chk文件并重启计算完成。
/ssadndp.exe SSAdNDP-UD.inp SSAdNDP.chk
直到最后P(NAO)中剩余的电子数(<<2)不能再组成一个多中心键为止。
按照文章的思路,最后我找到三个2c-2e,NO=1.72。
一个6c-2e,NO=1.48。剩余电子1.21,我认为没必要继续往下找了,原文中还继续找到了8c-2e键。
第七步:可视化轨道,用VESTA做图。
./visual.exe vis_gs_2c-bonds.out 50 50 50
生成的gs_2c-bond_1.cube和lattice_vec.cube文件按照手册说明在VESTA里作图即可。
转载请注明来源,欢迎对文章中的引用来源进行考证,欢迎指出任何有错误或不够清晰的表达。