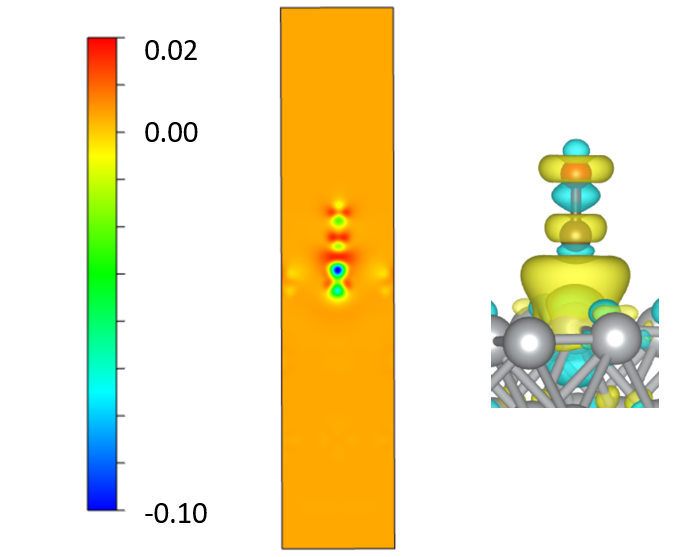
CONTCAR\(3) 1.00000000000000 4.9777998924000002 0.0000000000000000 0.0000000000000000 0.0000000000000000 4.9777998924000002 0.0000000000000000 0.0000000000000000 0.0000000000000000 23.7996006012000016 Ni C O 24 1 1 Selective dynamics Direct 0.0000000000000000 0.0000000000000000 0.2000000000000028 F F F 0.0000403141048793 0.0001267891084566 0.3465324639523857 T T T 0.0003869756759585 0.0005016061154492 0.4914092265927721 T T T 0.2500000000000000 0.2500000000000000 0.1260499910000021 F F F 0.2501509938760123 0.2501939027302242 0.2738045972087804 T T T 0.2510445350818244 0.2510358567191133 0.4214201499164574 T T T 0.5000000000000000 0.0000000000000000 0.2000000000000028 F F F 0.5001131027590162 0.0001793013434579 0.3476242737143451 T T T 0.5003637542315573 0.9997315722502194 0.4921789851389491 T T T 0.7499999759999980 0.2500000000000000 0.1260499910000021 F F F 0.7499316556673818 0.2502046005629239 0.2738030513532408 T T T 0.7491890345597767 0.2510567755592561 0.4213836669568778 T T T 0.0000000000000000 0.5000000000000000 0.2000000000000028 F F F 0.0001477148529645 0.5002040345309098 0.3476407140178068 T T T 0.9998279455461869 0.5004850843478295 0.4921814441992751 T T T 0.2500000000000000 0.7499999759999980 0.1260499910000021 F F F 0.2501233421650753 0.7499923211510620 0.2738063435823861 T T T 0.2509685482545621 0.7492259105458245 0.4213662097234874 T T T 0.5000000000000000 0.5000000000000000 0.2000000000000028 F F F 0.5000095461819711 0.5000093692680139 0.3475763670314436 T T T 0.4997626166386695 0.4997469098518152 0.4975764390870765 T T T 0.7499999759999980 0.7499999759999980 0.1260499910000021 F F F 0.7499481468791345 0.7499759622207023 0.2737998574575187 T T T 0.7493532326515719 0.7494160592036465 0.4213303989337192 T T T 0.5053523115939882 0.5076925776123602 0.5706792117914432 T T T 0.5089497223101063 0.5124605842843479 0.6195520236066443 T T T
314 ======================= File Options ============================ Input the Names of Charge/Potential Files with Space: (e.g., to get AB-A-B, type: ~/AB/CHGCAR ./A/CHGCAR ../B/CHGCAR) (e.g., to get A-B, type: ~/A/CHGCAR ./B/CHGCAR)
------------>> ./CHGCAR ./co/CHGCAR ./slab/CHGCAR
-->> (01) Reading Structural Parameters from ./CHGCAR File... -->> (02) Reading Charge Density From ./CHGCAR File... -->> (03) Reading Structural Parameters from ./co/CHGCAR File... -->> (04) Reading Charge Density From ./co/CHGCAR File... -->> (05) Reading Structural Parameters from ./slab/CHGCAR File... -->> (06) Reading Charge Density From ./slab/CHGCAR File... -->> (07) Written CHGDIFF.vasp File!
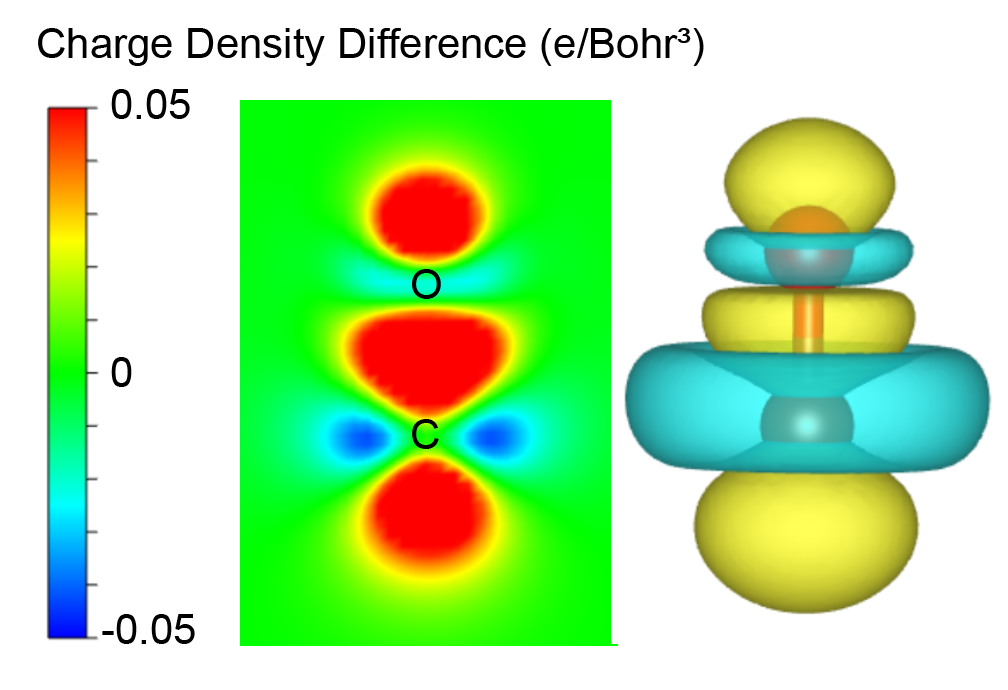
314 ======================= File Options ============================ Input the Names of Charge/Potential Files with Space: (e.g., to get AB-A-B, type: ~/AB/CHGCAR ./A/CHGCAR ../B/CHGCAR) (e.g., to get A-B, type: ~/A/CHGCAR ./B/CHGCAR)
------------>> ./CHGCAR ./deform/CHGCAR
-->> (01) Reading Structural Parameters from ./CHGCAR File... -->> (02) Reading Charge Density From ./CHGCAR File... -->> (03) Reading Structural Parameters from ./deform/CHGCAR File... -->> (04) Reading Charge Density From ./deform/CHGCAR File... -->> (05) Written CHGDIFF.vasp File!
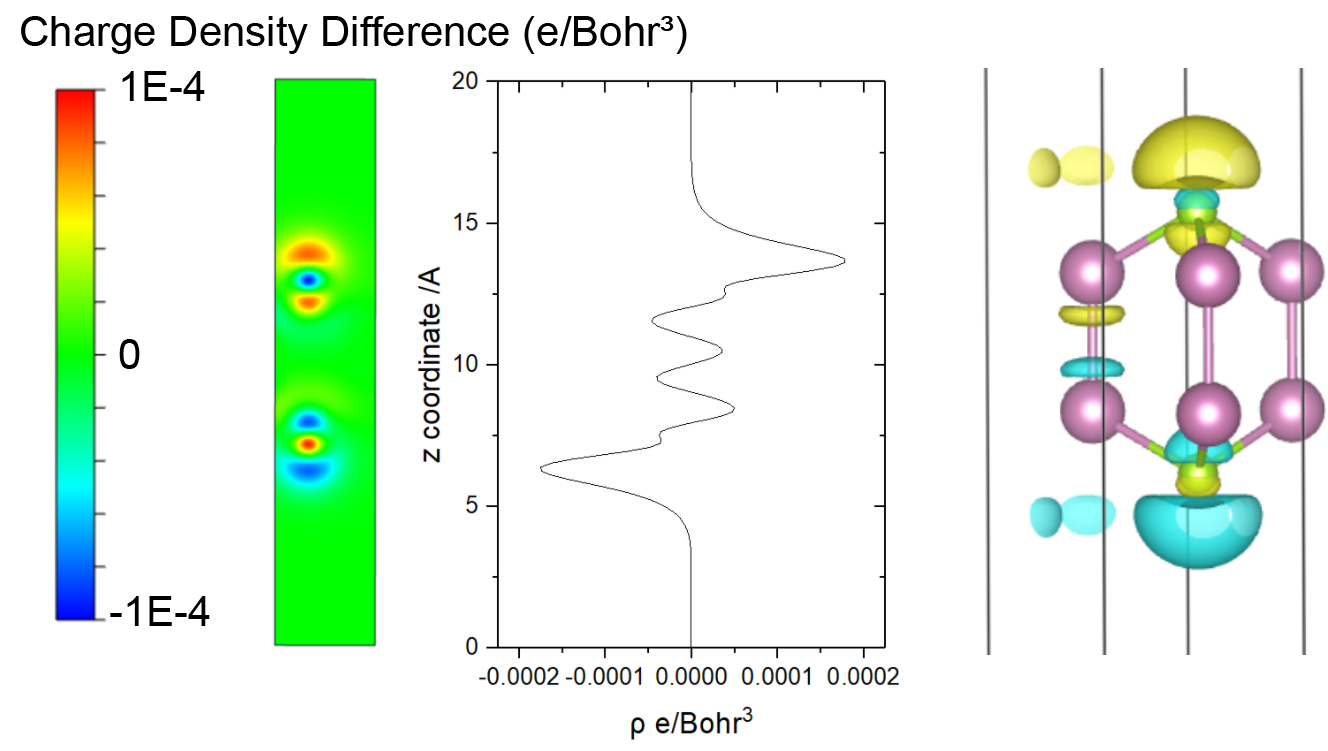
314 ======================= File Options ============================ Input the Names of Charge/Potential Files with Space: (e.g., to get AB-A-B, type: ~/AB/CHGCAR ./A/CHGCAR ../B/CHGCAR) (e.g., to get A-B, type: ~/A/CHGCAR ./B/CHGCAR)
------------>> ./CHGCAR ./noele/CHGCAR
-->> (01) Reading Structural Parameters from ./CHGCAR File... -->> (02) Reading Charge Density From ./CHGCAR File... -->> (03) Reading Structural Parameters from ./noele/CHGCAR File... -->> (04) Reading Charge Density From ./noele/CHGCAR File... -->> (05) Written CHGDIFF.vasp File!