from pymatgen import Structure, Lattice, MPRester, Molecule from pymatgen.analysis.adsorption import * from pymatgen.core.surface import generate_all_slabs from pymatgen.symmetry.analyzer import SpacegroupAnalyzer from matplotlib import pyplot as plt from pymatgen.ext.matproj import MPRester from pymatgen.io.vasp.inputs import Poscar
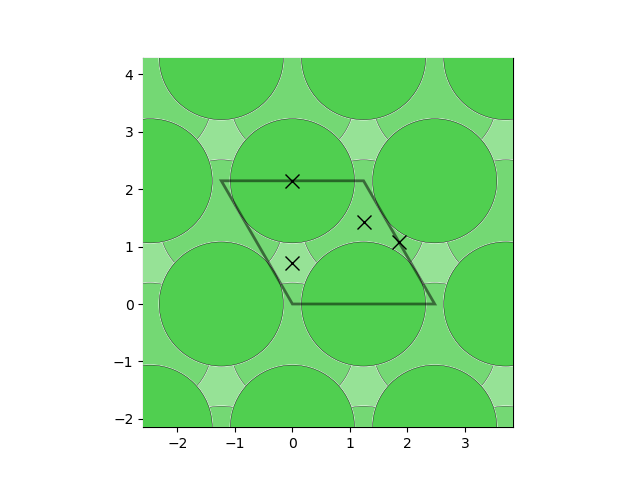
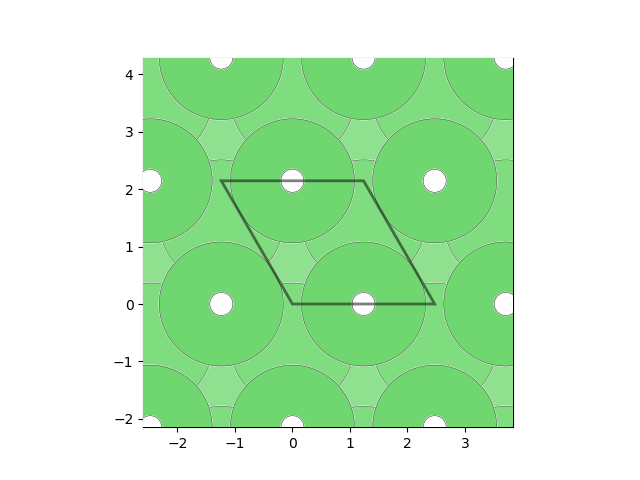
fcc_ni = Structure.from_spacegroup("Fm-3m", Lattice.cubic(3.5), ["Ni", "Ni"], [[0, 0, 0], [0.5, 0.5, 0.5]]) slabs = generate_all_slabs(fcc_ni, max_index=1, min_slab_size=8.0, min_vacuum_size=15.0) ni_111 = [slab for slab in slabs if slab.miller_index==(1,1,1)][0]
# Import statements from pymatgen import Structure, Lattice, MPRester, Molecule from pymatgen.analysis.adsorption import * from pymatgen.core.surface import generate_all_slabs from pymatgen.symmetry.analyzer import SpacegroupAnalyzer from matplotlib import pyplot as plt from pymatgen.ext.matproj import MPRester from pymatgen.io.vasp.inputs import Poscar # Note that you must provide your own API Key, which can # be accessed via the Dashboard at materialsproject.org mpr = MPRester('yourPrivateKey')
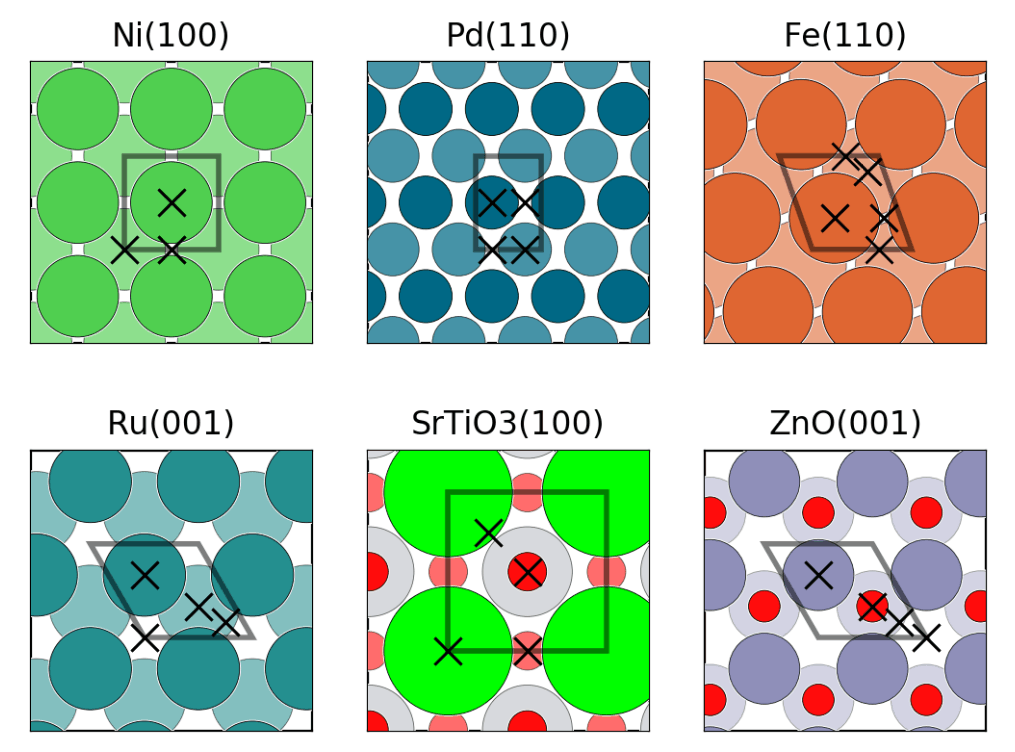
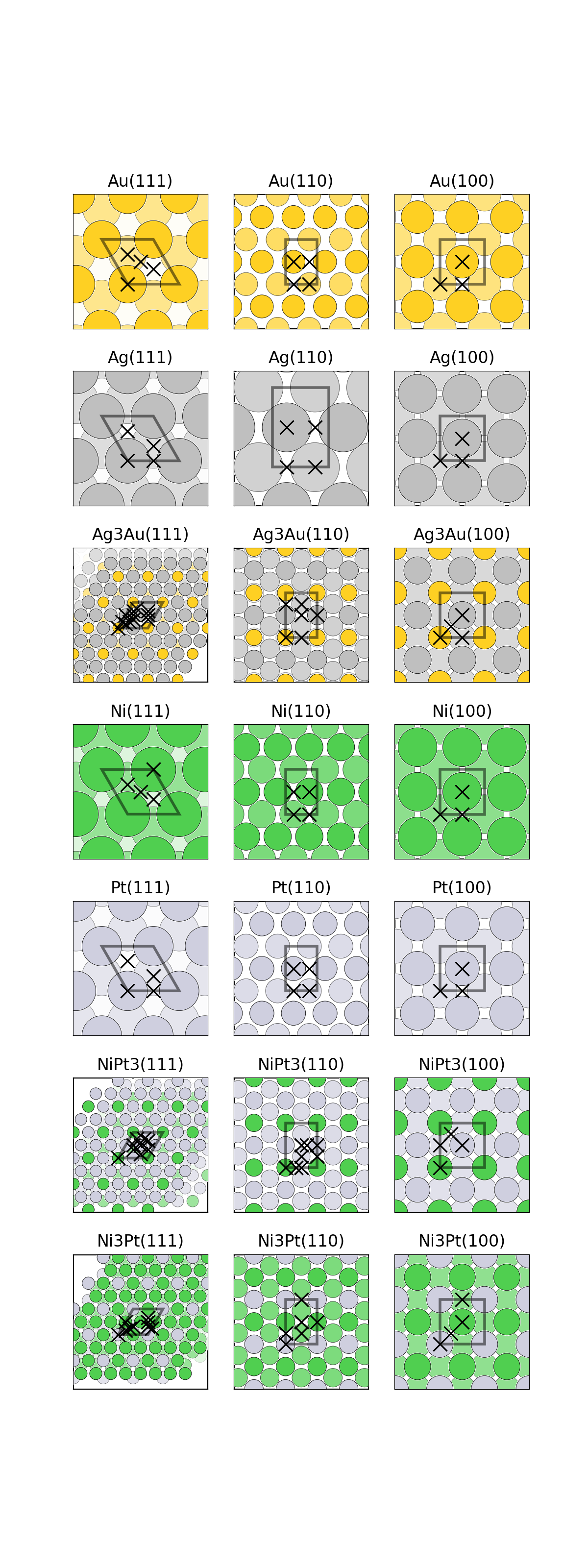
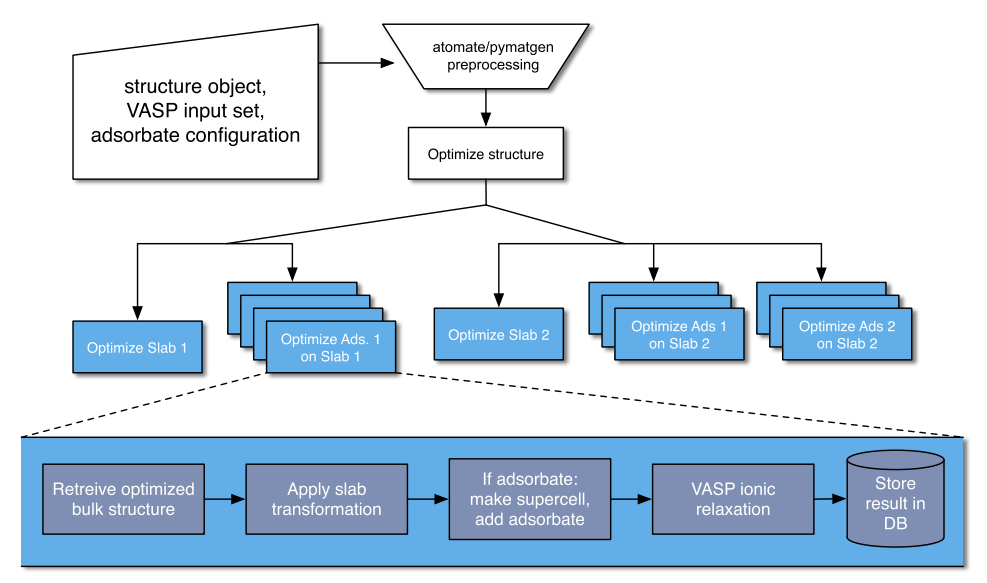
fig = plt.figure() axes = [fig.add_subplot(2, 3, i) for i in range(1, 7)] mats = {"mp-23":(1, 0, 0), # FCC Ni "mp-2":(1, 1, 0), # FCC Au "mp-13":(1, 1, 0), # BCC Fe "mp-33":(0, 0, 1), # HCP Ru "mp-5229":(1, 0, 0), # Cubic SrTiO3 "mp-2133":(0, 0, 1)} # Wurtzite ZnO for n, (mp_id, m_index) in enumerate(mats.items()): struct = mpr.get_structure_by_material_id(mp_id) struct = SpacegroupAnalyzer(struct).get_conventional_standard_structure() slabs = generate_all_slabs(struct, 1, 5.0, 2.0, center_slab=True) slab_dict = {slab.miller_index:slab for slab in slabs} asf = AdsorbateSiteFinder.from_bulk_and_miller(struct, m_index) plot_slab(asf.slab, axes[n]) ads_sites = asf.find_adsorption_sites() sop = get_rot(asf.slab) ads_sites = [sop.operate(ads_site)[:2].tolist() for ads_site in ads_sites] axes[n].plot(*zip(*ads_sites), color=’k’, marker=’x’,markersize=10, mew=1,\ linestyle=’’, zorder=10000) mi_string = "".join([str(i) for i in m_index]) axes[n].set_title("{}({})".format(struct.composition.reduced_formula, mi_string)) axes[n].set_xticks([]) axes[n].set_yticks([])